

NEURAL CORRELATES AND THE DOPAMINE HYPOTHESIS

PLEASE NOTE:
Biochemical theories do not compete with genetic theories. They can be complementary; for example, genes could cause a person to have hypodopaminergic or hypadopaminergic systems.
This topic focuses on the dopamine hypothesis as the primary example of neural correlates. That said, it is vital to be aware that dopamine is not the only neural correlate for schizophrenia. For example, research has found that people with schizophrenia often have enlarged brain ventricles. These are fluid-filled spaces in the brain, and their enlargement suggests a loss of brain tissue linked to negative symptoms and cognitive impairment. While this is a well-documented neural correlate, this nor other neural correlates are not discussed at length in this thread.
DOPAMINE HYPOTHESIS KEYWORDS
AMPHETAMINE/CANNABIS/COCAINE PSYCHOSIS:
A condition in which high doses or chronic use of amphetamines (stimulants) produce symptoms that resemble schizophrenia, including hallucinations and delusions. This phenomenon supports the dopamine hypothesis by showing that excessive dopamine release can induce psychotic symptoms in otherwise healthy individuals.
AGONISTS / STIMULANTS:
Drugs that increase the activity of neurotransmitters in the brain. They may act by mimicking the neurotransmitter, increasing its release, or blocking its breakdown. Depending on the neurotransmitter system targeted, agonists may produce stimulant, euphoric, or sedative effects.
LEGAL AGONISTS (PSYCHOTROPICS):
L-DOPA – Dopamine precursor used to treat Parkinson’s disease
Methadone – Opioid receptor agonist used in addiction treatment
Prozac (fluoxetine) – Selective serotonin reuptake inhibitor (SSRI); enhances serotonin signalling
Valium (diazepam) – GABA enhancer; increases the calming effect of GABA
ILLEGAL AGONISTS (STREET DRUGS):
Cocaine – Blocks dopamine reuptake; increases dopamine signalling
Amphetamines (speed) – Increases release of dopamine and norepinephrine
MDMA (ecstasy) – Increases serotonin, dopamine, and norepinephrine
Cannabis (THC) – CB1 receptor agonist; indirectly affects dopamine
Heroin – Opioid receptor agonist; indirectly increases dopamine in reward pathways
Crack – Smokable form of cocaine with a rapid dopamine-enhancing effect
ANTAGONISTS / BLOCKERS:
Drugs that reduce the activity of neurotransmitters by blocking their receptors. Dopamine antagonists are used as antipsychotics (psychotropics) and are primarily prescribed to reduce the positive symptoms of schizophrenia, such as hallucinations and delusions.
LEGAL DOPAMINE ANTAGONISTS (PSYCHOTROPICS / ANTIPSYCHOTICS):
Chlorpromazine (Thorazine) – D2 receptor blocker
Haloperidol – Potent D2 receptor antagonist
Risperidone – Atypical antipsychotic; blocks both dopamine and serotonin receptors
Clozapine – Atypical antipsychotic; targets dopamine and serotonin systems
Note: There are no illegal drugs known to act primarily as dopamine antagonists. Antagonist effects are typically found in legally regulated medications, not recreational street drugs. But there are illegal drugs that block other neurotransmitters.
ILLEGAL BLOCKERS (NON-DOPAMINE):
PCP (Phencyclidine) – NMDA glutamate receptor antagonist
Ketamine – NMDA glutamate receptor antagonist
Scopolamine (Datura, Devil’s Breath) – Acetylcholine receptor antagonist
Diphenhydramine (DPH, Benadryl abuse) – Histamine (H1) and acetylcholine receptor antagonist
ANTIPSYCHOTICS / NEUROLEPTICS:
Antipsychotics (also known as neuroleptics) are drugs used to treat psychosis, including core symptoms of schizophrenia such as hallucinations, delusions, and disorganised thinking. The term neuroleptic traditionally refers to the first-generation (or typical) antipsychotics which emerged in the 1950s. These drugs often cause strong sedation and motor side effects (e.g. rigidity, tremors) as they block dopamine D2 receptors.
The broader term antipsychotic includes both these older drugs and the second-generation (or atypical) antipsychotics, which emerged later and tend to act on both dopamine and serotonin receptors. Atypicals are generally associated with fewer motor side effects but may have metabolic risks.
In contemporary research and clinical use, antipsychotic is the preferred term
ANTIHISTAMINE: A drug that blocks histamine receptors to reduce allergic reactions. Some antihistamines, like promethazine, also have sedative and tranquillising effects.
CALCIUM (Ca²⁺): A vital ion in the brain that regulates neurotransmitter release, neuronal excitability, and synaptic plasticity. Calcium plays a crucial role in signal transmission within neurons and interacts with glutamate through the activation of NMDA receptors. Disruptions in calcium signalling are associated with neuropsychiatric disorders, including schizophrenia.
CHLORPROMAZINE: Chlorpromazine is a phenothiazine antipsychotic developed initially as an antihistamine. It alleviates psychotic symptoms by blocking dopamine D2 receptors in the brain. It is marketed under the brand name Thorazine.
Drugs are typically known by two types of names: a generic (or chemical) name and a brand (or trade) name. The generic name, such as chlorpromazine, refers to the active ingredient and is used universally in scientific and clinical contexts. The brand name—such as Thorazine—is created by pharmaceutical companies for marketing purposes and may vary across countries.
When discussing psychological research or clinical studies, the generic name should be used, as it ensures clarity, avoids regional variation, and maintains academic consistency.
ALL ABOUT DOPAMINE
DOPAMINE:
A neurotransmitter that plays a key role in movement, motivation, reward, attention, learning, emotional regulation, and psychosis. It is central to the dopamine hypothesis of schizophrenia, which links excessive dopamine activity to positive symptoms (hallucinations and delusions) and reduced activity to negative symptoms (apathy, withdrawal). Dopamine also regulates behaviour, mood, and cognitive functions, such as decision-making, reinforcement learning, and the pursuit of goals and pleasure.
DOPAMINE RECEPTORS ARE DIVIDED INTO TWO PROMINENT FAMILIES:
D1-LIKE RECEPTORS (D1, D5): Generally stimulate neuron activity (excitatory effects).
D2-LIKE RECEPTORS (D2, D3, D4): Generally inhibit neuron activity (inhibitory effects).
These effects vary depending on brain region and neural pathway.
DIFFERENT DOPAMINE RECEPTORS ARE DISTRIBUTED IN SPECIFIC AREAS OF THE BRAIN, ENABLING THEM TO CONTROL DISTINCT FUNCTIONS:
D1 AND D2 RECEPTORS: Located in the basal ganglia (especially the striatum); both are essential for motor control, with D1 generally excitatory and D2 inhibitory in function.
D2 RECEPTORS: Also concentrated in the mesolimbic pathway, where they are implicated in psychotic symptoms (e.g. hallucinations) and reward processing.
D3 RECEPTORS: Primarily located in the limbic system, associated with emotional regulation, motivation, and reward sensitivity.
D4 RECEPTORS: Found in the frontal cortex, particularly involved in attention, impulse control, and decision-making; has been linked to disorders like ADHD and schizophrenia.
D5 RECEPTORS: Distributed in the hippocampus and hypothalamus; associated with learning, memory, and modulation of neuronal excitability.
DOPAMINE RECEPTORS SHAPE HOW WE RESPOND TO REWARDING EXPERIENCES. FOR EXAMPLE:
Imagine receiving a message from someone you like. Your phone lights up — dopamine is released. But how you feel, interpret, respond to, and remember that moment depends on the type and activity of specific dopamine receptors in different brain regions.
D1 RECEPTORS – MOTIVATION AND REINFORCEMENT
D1 receptors increase activity in brain circuits that support goal-directed behaviour and reward-based learning. They help the brain connect actions with positive outcomes.
In this example: You interpret the message as a positive outcome of something you did — confidence, timing, effort. D1 receptors strengthen the link between your behaviour and the reward, making it more likely you’ll repeat it.
Too little D1: The message doesn’t feel rewarding. You don’t feel motivated or energised, and you fail to learn from the experience.
Too much D1: You may become overcommitted to a specific goal or behaviour, repeating it even when it’s no longer rewarding. Overactivation of D1 is associated with rigid reinforcement, but not directly with psychosis.
D2 RECEPTORS – SALIENCE FILTERING AND EMOTIONAL RESTRAINT
D2 receptors regulate how much emotional or motivational weight is assigned to stimuli. They help the brain distinguish between what is meaningful and what isn’t — keeping responses proportionate.
In this example: D2 receptors ensure the message registers as meaningful, but not overwhelming. They help keep your emotional response in check.
Too much D2 activity (especially in the mesolimbic pathway): You may overinterpret the message — reading excessive meaning into it, or believing it confirms something important or personal. This is known as aberrant salience, and is a key mechanism in psychosis (e.g. delusions, paranoia).
Too little D2 activity (especially in the frontal cortex): The message may feel emotionally flat or irrelevant. You may struggle to care or respond — linked to negative symptoms such as apathy and avolition.
D3 RECEPTORS – ANTICIPATION AND EMOTIONAL CHARGE
D3 receptors are highly sensitive to reward cues and play a major role in anticipation, emotional responsiveness, and craving.
In this example: Even before you read the message, you feel a surge of anticipation and emotional arousal. D3 receptors respond to the cue — the notification tone or name on your screen.
Too little D3: Anticipation is dulled. Reward cues don’t evoke excitement, and even promising experiences feel emotionally flat.
Too much D3: You may become fixated on cues — repeatedly checking your phone, overinterpreting signs, or developing craving-like responses. D3 overactivity is linked to addiction, compulsivity, and relapse risk.
D4 RECEPTORS – ATTENTION AND RESPONSE CONTROL
D4 receptors help regulate attention, cognitive flexibility, and impulse control. They support the ability to assess situations, weigh responses, and shift focus appropriately.
In this example: You read the message, pause, and think about how to respond. D4 helps you process the tone, avoid reacting impulsively, and tailor your reply.
Too little D4: You may respond without thinking, misread tone, or get stuck thinking about the message afterwards. D4 deficits are linked to impulsivity, distractibility, and ADHD.
Too much D4: You may overanalyse the message, second-guess your response, or become stuck in indecision and self-monitoring.
D5 RECEPTORS – EMOTIONAL LEARNING AND MEMORY
D5 receptors support the encoding of emotionally significant experiences. They help the brain learn from outcomes and shape future behaviour based on past emotional events.
In this example: D5 receptors help you store the emotional and motivational aspects of the message — what it meant, how it felt, and what led to it.
Too little D5: The experience may be poorly encoded. You might not learn from it, or fail to recall how it felt later.
Too much D5: The memory may become over-consolidated — excessively vivid or emotionally charged — potentially leading to rumination, emotional fixation, or unrealistic expectations
GLUTAMATE: The brain’s most abundant excitatory neurotransmitter — often referred to as the “on switch.” It supports synaptic transmission, learning, and memory, and regulates other neurotransmitters, such as dopamine. Dysregulation of glutamate, especially at NMDA receptors, is linked to both positive and negative schizophrenia symptoms.
GLYCINE: An amino acid and co-agonist for NMDA receptors. Glycine enhances NMDA function and may help treat cognitive and negative symptoms of schizophrenia when levels are deficient.
HYPER- AND HYPO- (NEUROTRANSMITTER IMBALANCES): The prefixes hyper- and hypo- are often confused due to their similar sounds but have opposite meanings. Hyper- means excessive, overactive, or above normal, as seen in words like hyperbole (extreme exaggeration) and hyperactive (excessively active). In contrast, hypo- means low, underactive, or below normal, as in hypoglycemia (low blood sugar) and hyposensitivity (reduced sensitivity).
When referring to neurotransmitters, a hyperdopaminergic system occurs when excessive dopamine is released into synapses, leading to the overstimulation of neurons and exaggerated responses. This is seen in schizophrenia’s positive symptoms, such as hallucinations and delusions, where excessive dopamine activity in the mesolimbic pathway disrupts normal perception and thought processing.
Conversely, a hypodopaminergic system indicates insufficient dopamine activity, causing reduced neuronal stimulation. In schizophrenia, low dopamine levels in the mesocortical pathway contribute to negative symptoms, such as apathy, lack of motivation, and social withdrawal, due to the interactivity of dopamine-dependent cognitive and emotional functions.
Understanding the distinction between hyperdopaminergic and hypodopaminergic states helps explain why schizophrenia presents with both excessive and diminished mental activity, affecting different brain pathways in various ways.
NEURAL CORRELATES: Physical or functional changes in specific brain areas linked to observable symptoms. In schizophrenia, altered dopamine activity in the striatum, prefrontal cortex, or limbic system is associated with delusions, emotional flatness, or disorganised thinking.
NEUROTRANSMITTERS: Chemical messengers that allow neurons to communicate by crossing synapses and binding to receptors. Examples include dopamine, serotonin, glutamate, GABA, and acetylcholine.
NMDA RECEPTORS: A subtype of glutamate receptor essential for synaptic plasticity, memory, and learning. Dysfunction of NMDA receptor activity is central to the glutamate hypothesis of schizophrenia, helping to explain hallucinations, cognitive decline, and flattened emotional responses.
PARKINSON’S DISEASE:
A neurodegenerative disorder caused by dopamine depletion in the substantia nigra, a brain region critical for movement. Symptoms include tremors, rigidity, and slow, deliberate movements. Parkinson’s is often treated with dopamine-boosting drugs like L-DOPA. The link between dopamine loss and motor dysfunction in Parkinson’s helped researchers understand dopamine’s role in both movement and mental health.
PHENOTHIAZINE: A tricyclic chemical compound composed of carbon, nitrogen, and sulfur. Used as the base for a class of early antipsychotic drugs. Chlorpromazine is a phenothiazine derivative that blocks dopamine D2 receptors.
PROMAZINE:
A first-generation (typical) antipsychotic chemically related to chlorpromazine. While it shares the phenothiazine structure, promazine is generally less potent and is more commonly used for short-term sedation or management of agitation, particularly in non-psychotic patients. It is not usually a first-line treatment for the core symptoms of schizophrenia, such as hallucinations or delusions.
PROMETHAZINE:
A first-generation antihistamine with sedative and anticholinergic properties. It is chemically related to phenothiazines and was a precursor to the development of antipsychotic drugs like chlorpromazine. Though not used as an antipsychotic, its tranquillising effects influenced early psychiatric drug discovery.
PSYCHOTROPICS:
Psychotropics are substances that affect the mind, mood, perception, or behaviour by altering brain chemistry. In clinical settings, the term usually refers to legal pharmaceutical drugs developed and regulated by the medical industry (often referred to as Big Pharma). These include medications prescribed for mental health conditions such as depression, schizophrenia, anxiety disorders, and bipolar disorder. They typically act on neurotransmitters like dopamine, serotonin, GABA, or norepinephrine, functioning as agonists, antagonists, or modulators at receptor sites.
Examples of legal psychotropics:
Antipsychotics (e.g., chlorpromazine, risperidone)
Antidepressants (e.g., Prozac, sertraline)
Anxiolytics (e.g., diazepam, alprazolam)
Mood stabilisers (e.g., lithium)
Stimulants (e.g., methylphenidate, amphetamines for ADHD)
Illegal or controlled psychotropics — such as LSD, psilocybin, MDMA, or cannabis (in many jurisdictions) — also alter perception, mood, and cognition, but are not typically produced or regulated by pharmaceutical companies. While some are now being studied or used in clinical trials, their legal status remains restricted in many countries.
This distinction reflects not just legality, but also institutional control, regulatory status, and therapeutic intent
RESERPINE:
A naturally derived drug from Rauwolfia serpentina that depletes monoamines (dopamine, norepinephrine, and serotonin) by blocking their storage in synaptic vesicles (via VMAT2 inhibition). It was used in the 1950s to treat high blood pressure and psychosis, but it frequently caused severe depression and Parkinsonian side effects. These effects helped establish links between dopamine depletion, mood disorders, and motor control, contributing to the development of both the dopamine hypothesis of schizophrenia and models of Parkinson’s disease.
SEROTONIN: A neurotransmitter involved in regulating mood, sleep, appetite, and emotion. While primarily associated with depression, serotonin also modulates dopamine activity. Many second-generation antipsychotics (e.g., risperidone, clozapine) block both serotonin and dopamine receptors to improve outcomes for negative and cognitive symptoms.
THORAZINE: The trade name for chlorpromazine, introduced in 1952. It was the first widely used antipsychotic and marked the beginning of modern drug treatment for schizophrenia.
WHAT ARE NEURAL CORRELATES?
A neural correlate is a change in brain activity, function, or structure that is consistently linked to a specific behaviour or mental state. These differences may be caused by genes, disease, epigenetic factors, or environmental influences. For example, people who have difficulty producing speech often show damage to a region in the left frontal lobe known as Broca’s area. This suggests a strong relationship between that brain region and speech output. In clinical psychology, neural correlates are used to identify biological features of mental illness by linking brain abnormalities with patterns of symptoms. While correlations alone do not prove cause and effect, in many cases — especially where brain damage leads to a clear behavioural change — a causal relationship is strongly supported by converging evidence.
A neural correlate for schizophrenia is a physical difference in the brain that is linked to the symptoms of the disorder. It refers to specific, observable features in brain structure or activity that consistently occur in people with schizophrenia.
Research has identified many neural correlates for schizophrenia, meaning areas of brain damage or differences that correspond with schizophrenic behaviour. However, because this is a large area, AQA requires you to focus on the dopamine hypotheses as the neural correlate to discuss in your essay.
Dopamine dysregulation is one of the key neural correlates of schizophrenia. This theory suggests that excess dopamine activity in the mesolimbic pathway is linked to positive symptoms such as hallucinations and delusions. In contrast, reduced dopamine activity in the mesocortical pathway is associated with “negative” symptoms like avolition and speech poverty. This makes dopamine dysfunction a crucial biological explanation of schizophrenia.

BACKGROUND TO THE DOPAMINE HYPOTHESIS
PHENOTHIAZINE, ANTIHISTAMINES AND SURGICAL SHOCK (1940s)
Phenothiazine is a tricyclic chemical compound — meaning it has three connected rings — made up of carbon, nitrogen, and sulfur atoms. It’s not a drug itself but acts as a chemical base (or scaffold) used to build other drugs. Think of it like a car chassis: depending on how it’s modified, you can produce different kinds of therapeutic effects. For example, by altering the nucleus, scientists were able to create promethazine, an antihistamine with sedative effects, developed in the 1940s.
An antihistamine is a drug that blocks H1 histamine receptors, thereby reducing allergy symptoms such as sneezing, itching, and swelling. However, newer drugs like promethazine also have tranquilising properties. Laborit found that promethazine not only made patients more physiologically stable but also less anxious and emotionally stable. Promethazine blurred the line between physiological sedation and emotional modulation — something that hadn’t been pharmacologically separated before.
By 1949, French military surgeon Henri Laborit had begun using promethazine during surgery to prevent surgical shock — a dangerous drop in blood pressure caused by trauma or anaesthesia. Impressed by its calming effects, researchers at Rhône-Poulenc modified the molecule further, eventually synthesising chlorpromazine in 1950. Initially used as a pre-anaesthetic, chlorpromazine was soon found to reduce agitation, hallucinations, and delusions in psychiatric patients without fully sedating them. This marked the beginning of modern antipsychotic medication.
PSYCHIATRIC TESTING OF CHLORPROMAZINE (1951-1952)
Laborit’s findings caught the attention of psychiatrists Jean Delay and Pierre Deniker, who speculated that a drug that induced emotional detachment might help patients with severe psychiatric agitation.
In 1951, preliminary testing began on agitated psychiatric patients using chlorpromazine.
1952: Delay and Deniker expanded trials to schizophrenia patients at Sainte-Anne Hospital in Paris.
The results were unexpected and groundbreaking:
Unlike traditional sedatives, which only made patients drowsy, chlorpromazine alleviated psychotic symptoms by actively reducing delusions, hallucinations, and agitation.
Because of its unprecedented effectiveness in psychiatric trials, chlorpromazine (later marketed as Thorazine) was officially introduced as the first neuroleptic (first generation of antipsychotics, also known as typical or traditional antipsychotics) in 1952.
This discovery marked the beginning of modern pharmacological treatment for schizophrenia and established chlorpromazine as the first typical antipsychotic drug.
LINKING CHLORPROMAZINE TO SCHIZOPHRENIA
When chlorpromazine (later marketed as Thorazine) was introduced in the early 1950s, it was immediately clear that it produced effects unlike traditional sedatives. Barbiturates simply suppressed consciousness and induced sleep, whereas chlorpromazine reduced hallucinations, delusions, and agitation while leaving patients alert. Clinicians recognised that it worked differently — that it was doing something more specific than just calming or sedating — but they had no understanding of how.
At the time, the mechanisms of psychiatric drugs were a mystery. The concept of neurotransmitters was only just emerging, and the role of brain chemistry in mental illness was not yet part of scientific thinking. Terms like “calming the nerves” or “reducing stimulation” were placeholders for processes no one could yet describe. Neuroscience was onky just emerging, and with no clear theory of the brain communicated internally, observations remained disconnected from explanation.
THE DISCOVERY OF NEUROTRANSMITTERS
The first neurotransmitter, acetylcholine, was identified chemically in 1914 by Henry Dale, but it was Otto Loewi who first demonstrated its physiological function. In 1921, Loewi conducted a famous experiment using two frog hearts to show that nerve impulses could be transmitted chemically. He called the mysterious substance "Vagusstoff", which later turned out to be acetylcholine. This experiment provided the first direct evidence that neurons communicate via chemical messengers — a revolutionary finding at the time.
Even so, it took decades for scientists to identify other neurotransmitters. Dopamine, for example, was not recognised as a neurotransmitter until the late 1950s, when Swedish scientist Arvid Carlsson demonstrated its role in voluntary movement. At the time, dopamine was believed to be merely a chemical step in the synthesis of noradrenaline and not functionally crucial in its own right.
In the early 1950s, the drug reserpine — derived from the plant Rauwolfia serpentina — was introduced as a treatment for high blood pressure and agitation. However, it soon became clear that reserpine had unusual side effects: it caused emotional blunting, depression-like symptoms, and Parkinsonian movement problems in both humans and animals. These unexpected effects led researchers, such as Carlsson, to investigate whether reserpine worked by depleting key brain chemicals — and whether reversing that depletion could restore normal function.
Carlsson used reserpine to deplete dopamine in the brains of rabbits, which led to severe motor impairments. He then administered L-DOPA — a naturally occurring chemical precursor to dopamine — and found that regular movement was restored. This demonstrated that dopamine was not merely a biochemical intermediate but a neurotransmitter in its own right, playing an independent and essential role in initiating movement.
Despite these discoveries, neuroscience was still in its infancy. The notion that psychiatric symptoms could result from disruptions in neurotransmitter systems or that cognition itself might depend on chemical signalling between neurons — had not yet been established.
It was only in the 1960s that scientists began to connect dopamine imbalances to schizophrenia, leading to the first version of the dopamine hypothesis, now known as the Original Dopamine Hypothesis.
THE BIRTH OF THE ORIGINAL DOPAMINE HYPOTHESIS
Once chlorpromazine was shown to reduce psychotic symptoms, researchers began investigating the specific neurochemical changes it produced in the brain. This line of inquiry led to a series of findings that would ultimately implicate dopamine dysfunction in schizophrenia.
One early — but ultimately unsupported — hypothesis was that chlorpromazine acted on noradrenaline, a neurotransmitter associated with arousal and stress. However, by the early 1960s, researchers observed that high doses of amphetamines could induce symptoms such as paranoia, hallucinations, and delusions, closely resembling those seen in schizophrenia. These effects were first noted in recreational users and later replicated under controlled psychiatric conditions. Although the exact mechanisms were still unclear, amphetamines were known to increase dopamine activity, raising the possibility that excess dopamine might contribute to psychosis.
Around the same time, studies found that chlorpromazine and other chemically related antipsychotics and neuroleptics, including haloperidol, synthesised in 1958 — all shared a key pharmacological feature: they blocked dopamine receptors. The observation that some drugs could increase dopamine and trigger psychosis while others could block dopamine and alleviate it pointed to dopamine overactivity as a possible underlying factor in schizophrenia.
The dopamine hypothesis of schizophrenia was formally proposed by Jacques van Rossum in 1967, who suggested that schizophrenia was caused by dopamine dysregulation. This proposal marked a turning point in psychiatry and led to decades of research into dopamine’s role in the pathophysiology of schizophrenia.
HOW DOPAMINE CONTRIBUTES TO SCHIZOPHRENIA
Dopamine plays a crucial role in attention, motivation, and the perception of essential stimuli. It helps the brain decide what is relevant and what can be ignored, ensuring that a person stays focused on meaningful information rather than being overwhelmed by every minor detail in their environment.
In schizophrenia, this mechanism breaks down. Excess dopamine — particularly in the mesolimbic system — causes the brain to assign excessive importance to otherwise irrelevant stimuli. This phenomenon, known as aberrant salience, means that random thoughts, external events, or background sensations may feel unusually significant or emotionally charged. The system that normally ranks experiences by priority becomes flooded. Attention is drawn indiscriminately, and meaning is projected where none exists.
THE EFFECT OF EXCESS DOPAMINE ON THINKING AND LANGUAGE
The inability to suppress irrelevant information underpins many of the disordered thought patterns seen in schizophrenia:
Loose associations (also known as derailment): ideas are linked by weak or tangential connections. A person may shift topic mid-sentence, responding to a word, image, or sound that captures their attention without conscious intent.
Knight’s move thinking: a more extreme form of derailment, where thoughts leap unpredictably from one idea to another, without logical progression. Because no clear hierarchy of relevance exists, every thought feels equally compelling.
Clang associations: speech becomes driven by the sound of words — rhyme, rhythm, or alliteration — rather than their meaning. Phrases like “tight, night, fright, light” may seem meaningful, despite lacking semantic coherence.
These patterns reflect a deeper disruption in semantic integration. Rather than processing a sentence as a meaningful whole, the individual may latch onto a single word or syllable, treating it as if it carries hidden significance. Fragments of language become focal points, while context is ignored. The mind cannot decide what to filter out, so irrelevant stimuli are granted the same weight as meaningful ones.
HOW DOPAMINE LEADS TO DELUSIONS AND HALLUCINATIONS
When irrelevant information feels necessary, the brain tries to make sense of it. This is one of the reasons why schizophrenia is often accompanied by delusions of reference, where a person believes that unrelated events are personally significant.
For example, a person with schizophrenia might believe that a newsreader on television is sending them secret messages or that random strangers on the street are discussing them. This happens because excess dopamine makes neutral stimuli feel deeply meaningful.
This misattribution of significance can also lead to:
Paranoid behaviour – a belief that others are watching, judging, or planning against them.
Persecutory delusions – the conviction that one is being targeted, spied on, or conspired against.
Dopamine also plays a role in the distinction between internal and external experiences. A spontaneous thought may arise — triggered by an irrelevant stimulus but without a clear cause. The person may then attribute the thought to an external source. If a thought disappears, it may feel stolen. If inner speech is no longer recognised as self-generated, it may be experienced as an external voice — giving rise to auditory hallucinations.
These symptoms stem from a breakdown in self-monitoring: the brain’s ability to recognise its own activity. When combined with aberrant salience, the result is not just a misreading of the outside world, but a collapse in the sense of ownership over one’s own mind. Ordinary coincidences, such as a glance from a stranger or a phrase on television, may be experienced as coded messages. Delusions arise as the brain struggles to impose structure on a world in which relevance and origin can no longer be distinguished.
THE DOPAMINE HYPOTHESIS AND COGNITIVE INFLAMMATION
According to psychiatrist Shitij Kapur, dopamine is a biochemical fuel that amplifies specific ways of thinking. He argues that people who develop schizophrenia tend to jump to conclusions or interpret events in extreme ways. Excess dopamine inflames these cognitive patterns, pushing them into full psychosis.
Kapur explains:
“If you could test patients before they were psychotic, you’d probably find they tend to jump to conclusions or choose extreme explanations. When you add to this a biochemical fuel – excess dopamine – you inflame this way of thinking; that is what dopamine does.”
This suggests that dopamine dysregulation does not create new ways of thinking but amplifies already present tendencies, making them far more extreme and difficult to control.
SUMMARY
Dopamine regulates attention and motivation, helping to filter what is important from what is not.
Excess dopamine causes everything to seem meaningful, making it difficult to ignore irrelevant details.
This breakdown in filtering leads to psychotic symptoms such as delusions of reference, paranoia, and auditory hallucinations.
Kapur argues that dopamine fuels pre-existing cognitive tendencies, exaggerating them rather than creating them anew
RESEARCH FOR THE ORIGINAL DOPAMINE HYPOTHESIS
If a dopamine imbalance causes schizophrenia, there should be evidence of unusual dopamine activity in the brains of individuals with the disorder. Early and contemporary studies provide both support and critique for this hypothesis. Below is a summary of the key research evidence, from historical methods to modern imaging techniques.
The primary evidence used to support the dopamine hypothesis is the theory behind the success of typical and atypical antipsychotic drugs such as Thorazine (chlorpromazine), e.g., as they reduce dopamine firing, schizophrenia must be caused by excess dopamine. Activity. Moreover, not only do anti-psychotic drugs (dopamine antagonists) reduce positive symptoms (hallucinations, delusions) in type one schizophrenics, but when the same individuals are given drugs with a dopamine agonist, e.g., medications such as L-dopa that increase dopamine availability, then their symptoms became much worse.
RESEARCH ANALYSIS: ANTI-PSYCHOTICS
Also adding support to the theory is research on Parkinson’s sufferers and dopamine agonists. A lack of dopamine causes Parkinson's disease. As a result, Parkinson’s patients are treated with synthetic legal agonists to increase their dopamine availability (e.g., L-Dopa). However, if Parkinson’s patients are given high levels of L-dopa, they can suffer from positive symptoms, e.g., they can experience psychotic side effects which mimic the symptoms of schizophrenia. Conversely, Type 1 schizophrenics can suffer from Parkinson’s symptoms when on antipsychotic drugs.
RESEARCH ILEGAL STREET DRUGS
This conclusion is further supported by the research of drug addicts who use street drugs with dopamine agonist properties, such as LSD, cocaine, amphetamine, methamphetamine and other similar substances, as all illegal drugs dramatically increase the levels of dopamine in the brain. Indeed, drug addicts often have symptoms that resemble those present in psychosis, particularly after large doses or prolonged use. This type of addiction is usually referred to as "amphetamine psychosis" or "cocaine psychosis," which may produce experiences virtually indistinguishable from the positive symptoms associated with schizophrenia. In the early 1970s, several studies experimentally induced amphetamine psychosis in ordinary participants to better document the clinical pattern of schizophrenia.
It is also worth noting that when schizophrenics abuse street drugs (it should be noted that schizophrenia is comorbid with drug addiction), positive symptoms become much worse. For example, up to 75% of patients with schizophrenia have increased signs and symptoms of their psychosis when given moderate doses of amphetamine or other dopamine-like compounds/drugs, all given at doses that neuro-typical volunteers do not have any psychologically disturbing effects. Lastly, repeated exposure to high doses of antipsychotics (dopamine antagonists gradually reduced paranoid psychosis in these neurotypical participants. There are ethical issues with the above studies.
RESEARCH ANALYSIS ILLEGAL DRUGS
However, this type of research has also fallen out of favour with the scientific research community, as drug-induced psychosis is now thought to be qualitatively different from schizophrenia psychosis. Differences between the drug-induced states and the typical presentation of schizophrenia have now become more apparent, e.g., euphoria, alertness, and over-confidence. Some researchers believe these symptoms are more reminiscent of mania (manic side of bipolar depression) than schizophrenia.
POST-MORTEM STUDIES AND EARLY FINDINGS
Early studies often relied on post-mortem examinations to investigate dopamine receptors in the brains of individuals diagnosed with schizophrenia. Many of these studies reported increased dopamine receptor density, particularly in the striatum. However, this method is highly problematic for several reasons:
Real-Time Limitations: Post-mortem studies cannot measure live dopamine activity, which is crucial for understanding neurotransmitter function.
Medication Effects: Many individuals studied had taken antipsychotic drugs, which affect brain chemistry and receptor density. As a result, changes observed in post-mortem brains may reflect medication effects rather than the underlying biology of schizophrenia.
Generalisability Issues: Case studies from post-mortems often lack generalisability, as the brains studied may not represent the broader population of individuals with schizophrenia.
Diagnostic Ambiguity: Before the introduction of the DSM-5, schizophrenia samples often included individuals with bipolar disorder, catatonia, or a mix of negative and positive symptoms, which could skew findings.
These methodological issues explain why historical research findings on dopamine activity in schizophrenia were often inconsistent.
RESEARCH ON RATS
Chemical stimulation in rats is thought to support the dopamine hypothesis. In brief, rats are given dopamine antagonists (e.g., antipsychotic drugs such as chlorpromazine) and dopamine agonists (e.g., L-dopa, PCP and amphetamines). The behaviour that rats show when given agonists is thought to be like the positive and negative symptoms of schizophrenia in humans. For example, several animal models of schizophrenia are based on the experimental observation that phencyclidine (PCP) and amphetamines can induce behavioural changes that include locomotor hyperactivity, stereotyped behaviour, and social withdrawal (Murray and Horita 1979).
RESEARCH ANALYSIS RATS
Rats are not comparable to humans; not only do they not have a language, which is one of the key problem areas in schizophrenics, but psychologists do not have a viable way of assessing how disorganised or hallucinogenic a rat’s thoughts are whilst on L-dopa as they can’t ask a rat if it is hallucinating or delusional. Moreover, as the clinical interview is the only valid way of assessing schizophrenia in humans, one wonders how the researchers got over that problem when determining the rats ‘supposedly’ positive schizophrenic symptoms; schizophrenia may be unique only to humans.
On the other hand, rats and humans share many similarities, including comparable hormonal and nervous systems. Plus, we have almost identical hind, mid-, and forebrains. More importantly, rats and humans share similar mesolimbic systems, the pathway in which dopamine is processed, so the research would be valuable in assessing how antagonists and agonists affect dopamine receptors.
ANALYSIS SPECIFIC TO THE ORIGINAL DOPAMINE HYPOTHESIS.
An important observation is that schizophrenia is not the only disorder associated with dopamine; bipolar I, II (manic depression), schizoaffective disorder and acute transient psychosis are just some of the disorders related to this neurotransmitter. This means that excess dopamine might have more to do with psychosis than schizophrenia and is, therefore, only a partial explanation.
Also relevant is the fact that current research shows that one-third of individuals with schizophrenia do not respond to antipsychotics despite high levels of D2-receptor occupancy. In other words, they fit the criteria for the original dopamine hypothesis, but drugs that reduce dopamine activity do not alleviate their positive symptoms. This finding undermines the idea that excess dopamine causes schizophrenia. On the other hand, some health professionals believe that this result occurs when patients start chemotherapy too long after the start of their symptoms.
NEGATIVE SYMTOMS
More importantly, a large subset of schizophrenics do not suffer from positive symptoms and instead present with “negative” symptoms. In these cases, antipsychotics do not affect type-two negative symptoms whatsoever. Interestingly, if dopamine agonists, such as L-dopa, are administered, these symptoms can improve. Thus, a significant problem with the original dopamine hypothesis is that dopamine is not implemented in type 2 schizophrenia, where negative symptoms predominate.
Over the years, researchers recognised that the original dopamine hypothesis, which explained positive symptoms (e.g., hallucinations, delusions) as a result of increased dopamine activity, failed to account for negative symptoms(e.g., apathy, flattened affect, and social withdrawal) and cognitive deficits (e.g., poor working memory, attention problems).
But why did it take so long for researchers to recognise negative symptoms as a core feature of schizophrenia? And why did they initially assume blocking dopamine would improve all symptoms?
WHY WERE NEGATIVE SYMPTOMS INITIALLY IGNORED
For much of the 20th century, schizophrenia was primarily understood in terms of its positive symptoms, as these were the most obvious and disruptive. Early psychiatrists did recognise that some patients exhibited a form of progressive mental decline, including apathy and withdrawal. In 1899, Emil Kraepelin compared these symptoms to dementia, describing how specific individuals with schizophrenia seemed to deteriorate over time. In 1911, Eugen Bleuler introduced the term schizophrenia and identified features such as affective flattening, poverty of speech, and anhedonia as core symptoms of the disorder.
Despite these early observations, negative symptoms were largely ignored. They were not as dramatic as hallucinations and delusions, making them harder to study. Clinicians often assumed they were simply a reaction to psychosis rather than a distinct issue, believing that symptoms such as withdrawal and reduced speech were a byproduct of delusions or hallucinations. The focus of treatment was primarily on psychotic agitation, as this was considered the most pressing issue in hospitalised patients. As a result, positive symptoms took priority in both research and treatment. It was not until the 1970s and 1980s that negative symptoms were recognised as a distinct feature of schizophrenia, requiring separate investigation.
When antipsychotics were introduced in the 1950s, researchers initially believed they would improve all symptoms of schizophrenia, including negative ones. However, even as positive symptoms responded to treatment, many patients remained withdrawn and unmotivated, suggesting that negative symptoms were not merely a consequence of psychosis but an independent aspect of the disorder.
THE REFORMULATED DOPAMINE HYPOTHESIS
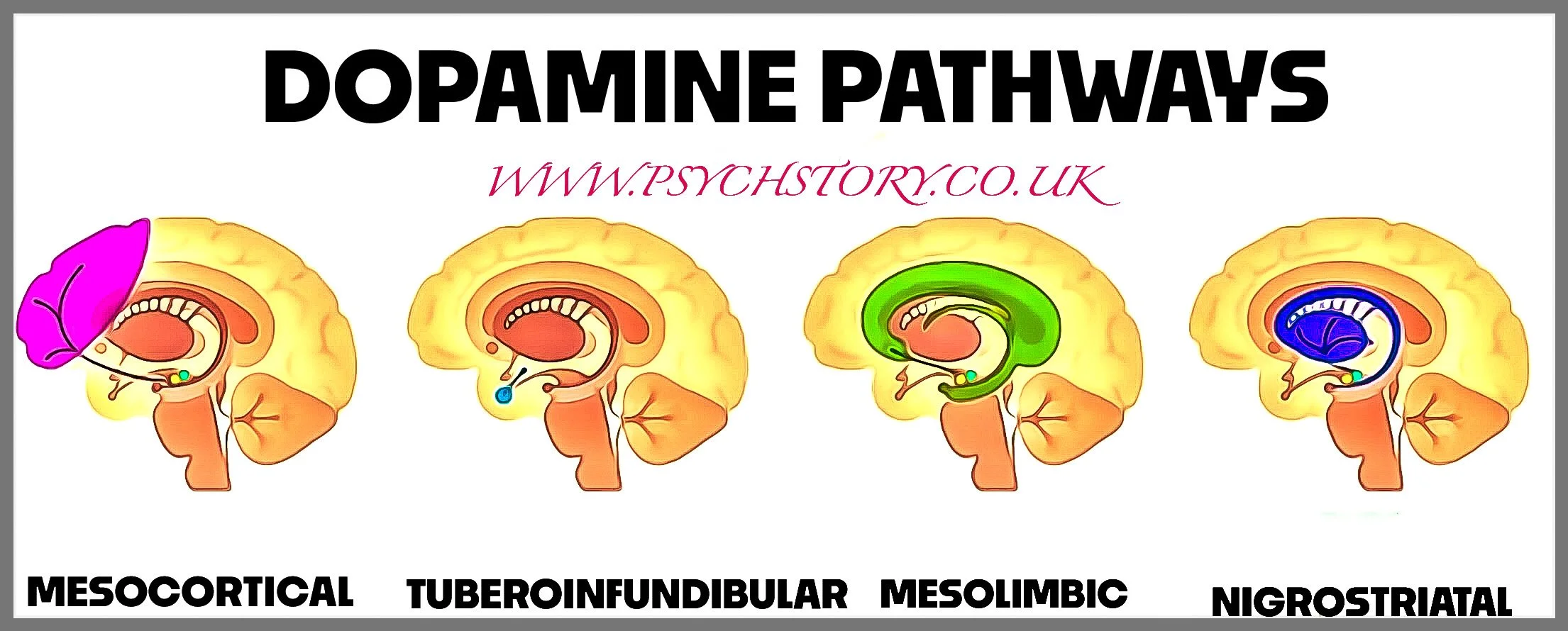
Further research has revealed that dopamine does not function uniformly throughout the brain. Instead, it operates in distinct pathways, each responsible for different aspects of behaviour. The mesolimbic pathway, which regulates emotion and reward, was found to be overactive, leading to positive symptoms such as hallucinations and delusions. Meanwhile, the mesocortical pathway, which is responsible for motivation, cognition, and decision-making, was found to be underactive, contributing to negative symptoms such as apathy, social withdrawal, and lack of motivation. This helped explain why schizophrenia could present with both excessive mental activity (psychosis) and a complete lack of drive and emotional expression.
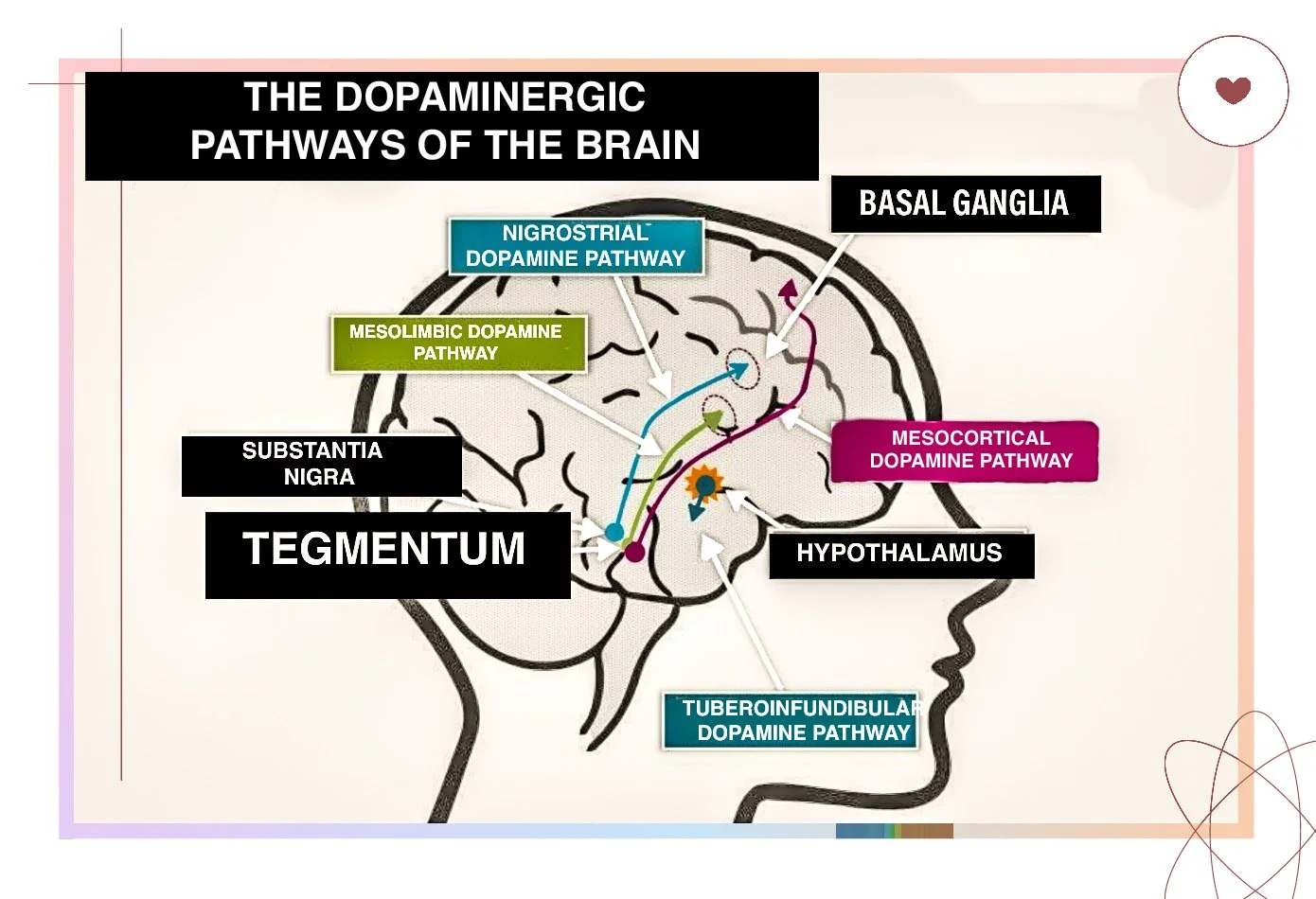
WHAT IS THE MESOLIMBIC SYSTEM?
The mesolimbic system is a subcortical dopamine pathway, meaning it is located beneath the cerebral cortex in deeper brain structures that control emotion, motivation, and reward processing. It connects two key brain areas:
The Ventral Tegmental Area (VTA) is where dopamine-producing neurons are located.
The Nucleus Accumbens receives dopamine signals and helps process reward, pleasure, and motivation.
When you experience something enjoyable, such as eating food or receiving praise, dopamine is released from the VTA and travels to the nucleus accumbens, reinforcing that behaviour. In schizophrenia, too much dopamine is released in this pathway, overstimulating the system and leading to positive symptoms.

HOW DOES DOPAMINE WORK? (D1 AND D2 RECEPTORS EXPLAINED)
Dopamine affects the brain by binding to dopamine receptors, which act like locks that dopamine "keys" can activate. These receptors come in two main types:
D1 receptors are excitatory, meaning they increase neuronal activity when stimulated. They are important for cognition, motivation, and decision-making, especially in the prefrontal cortex.
D2 receptors are inhibitory, meaning they reduce neuronal activity when dopamine binds to them. They play a key role in reward processing, motor control, and psychotic symptoms, particularly in subcortical regions like the mesolimbic system and the striatum.
WHY DOES TOO MUCH DOPAMINE IN THE MESOLIMBIC SYSTEM CAUSE POSITIVE SYMPTOMS?
D2 receptors in the mesolimbic system usually act as a brake, helping regulate emotional and sensory information. However, when dopamine levels are too high, these brakes become dysfunctional, leading to an overload of emotional and sensory information that the brain struggles to filter out. This overstimulation causes hallucinations, delusions, and thought disturbances.
The striatum, a part of the basal ganglia (another subcortical structure), also plays a role in the manifestation of positive symptoms. The associative striatum, involved in learning and cognitive processing, has been found to have excess dopamine activity in schizophrenia. When D2 receptors in the striatum are overstimulated, they interfere with the brain’s ability to organise thoughts, contributing to disorganised speech and erratic thinking.
WHY DOES TOO LITTLE DOPAMINE IN THE MESOCORTICAL SYSTEM CAUSE NEGATIVE SYMPTOMS?
In contrast, the mesocortical pathway, which connects the VTA to the prefrontal cortex, is underactive in schizophrenia. The prefrontal cortex is responsible for higher-order thinking, planning, motivation, and emotional regulation.
Dopamine in the prefrontal cortex primarily binds to D1 receptors, which are excitatory—meaning they increase neuronal activity and help maintain cognitive and motivational processes. When dopamine levels in this region are too low, D1 receptors receive insufficient stimulation, reducing neural activity and leading to:
Avolition (lack of motivation): Individuals struggle to initiate and sustain activities related to essential self-care.
Affective flattening: Emotional expression becomes diminished, leading to reduced facial expressions, a monotone voice, and social withdrawal.
Cognitive impairments: Attention, planning, and decision-making deficits make complex tasks challenging.

DIFFERENT TRAJECTORIES OF POSITIVE AND NEGATIVE SYMPTOMS
One key distinction between positive and negative symptoms is their progression over time. The difference in how these symptoms emerge, and progress is due to how dopamine dysfunction unfolds in different brain pathways.
WHY POSITIVE SYMPTOMS ARE ACUTE AND EPISODIC
Positive symptoms suddenly fluctuate because they are linked to dopamine surges in the mesolimbic system. Dopamine release in this region is influenced by external and internal triggers, such as stress, drug use, and even normal brain activity fluctuations. These factors can cause dopamine spikes, leading to episodes of psychosis that seem to appear rapidly.
This explains why hallucinations and delusions can be episodic—they often emerge during acute psychotic episodes, sometimes triggered by environmental factors or stress. Positive symptoms usually show rapid improvement with treatment because the mesolimbic pathway can be temporarily stabilised with dopamine-blocking medication.

WHY NEGATIVE SYMPTOMS DEVELOP SLOWLY AND ARE CHRONIC
In contrast, negative symptoms progress slowly and persist over time because they are associated with a gradual decline in dopamine function in the mesocortical pathway. Unlike the mesolimbic system, which reacts dynamically to changes in dopamine, the mesocortical system declines progressively, much like neurodegenerative diseases affect brain function over time.
This gradual decline in dopamine transmission to the prefrontal cortex means that motivational and cognitive impairments worsen over time rather than appearing suddenly. Since dopamine surges or episodic spikes do not drive this process, negative symptoms tend to be persistent and resistant to change, making them more long-lasting and difficult to treat compared to positive symptoms.
SUMMARY
The original dopamine hypothesis only explained positive symptoms, assuming all schizophrenia symptoms resulted from too much dopamine.
Negative symptoms were initially ignored or assumed to be a reaction to psychosis, not a separate dysfunction.
Early researchers did not yet understand that dopamine affects different brain regions differently, leading to false assumptions about treatment.
By the 1970s, it became clear that positive symptoms were caused by too much dopamine, while negative symptoms were linked to too little dopamine.
The reformulated dopamine hypothesis recognised that schizophrenia involves both hyperdopaminergic and hypodopaminergic activity, leading to a broader understanding of the disorder.
This updated model paved the way for research into cognition, motivation, and other neurotransmitters, moving beyond dopamine alone as the cause of schizophrenia.
RESEARCH ANALYSIS OF THE REFORMULATED DOPAMINE HYPOTHESIS
ADVANCES IN BRAIN IMAGING AND NEURAL CORRELATES
Modern techniques, such as PET (positron emission tomography) and fMRI (functional magnetic resonance imaging), have revolutionised the study of dopamine activity in living brains. These imaging methods provide real-time evidence of neurotransmitter function and structural changes in the brains of individuals with schizophrenia.
BRAIN IMAGING EVIDENCE FOR POSITIVE SYMPTOMS
Positive symptoms, such as hallucinations and delusions, have been linked to hyperdopaminergic activity in subcortical regions, particularly the mesolimbic system. Key findings include:
Increased Dopamine Receptors: Studies consistently show excess dopamine D2 receptors in the striatum, caudate nucleus, and amygdala.
Faster Dopamine Metabolism: Individuals with schizophrenia often exhibit increased dopamine turnover, reflecting faster synthesis and breakdown of dopamine in the brain.
Enhanced Dopamine Release: After taking amphetamines, which increase dopamine availability, individuals with schizophrenia release significantly more dopamine (particularly in the striatum) compared to neurotypical controls. This supports the link between dopamine overactivity and psychotic symptoms.
Auditory Hallucinations: Reduced activity in the superior temporal and anterior cingulate gyrus has been directly associated with auditory hallucinations. Patients experiencing these symptoms exhibit lower activation in these brain areas compared to healthy individuals.
RESEARCH STUDIES
Lindström et al. (1999) used PET scans to measure dopamine synthesis in people with schizophrenia and found increased dopamine production in the striatum compared to controls. This supports the idea that hyperdopaminergic activity in the striatum contributes to positive symptoms like hallucinations and delusions.
Howes et al. (2012) also used PET imaging. They found that people at high risk of developing schizophrenia had elevated dopamine synthesis in the striatum, suggesting that dopamine overactivity occurs before the onset of symptoms, supporting its role in causing psychosis rather than being a result of the disorder.
Amphetamine challenge studies (e.g., Laruelle et al., 1996) found that individuals with schizophrenia released more dopamine in response to amphetamines than control participants. Since amphetamines increase dopamine levels, this suggests that people with schizophrenia already have excess dopamine activity in the striatum, reinforcing the link between hyperdopaminergic and positive symptoms.
These findings suggest that hyperactivity in dopamine pathways and reduced activity in specific cortical areas act as neural correlates of positive symptoms.
BRAIN IMAGING EVIDENCE FOR NEGATIVE SYMPTOMS
Negative symptoms, such as avolition (loss of motivation) and social withdrawal, have been linked to hypodopaminergic activity in cortical regions. Key findings include:
Reduced Activity in the Ventral Striatum: The ventral striatum plays a key role in anticipating rewards and driving motivation. Abnormalities in this area are strongly linked to avolition.
Prefrontal Cortex and D1 Receptors: The under-functioning of D1 receptors in the prefrontal cortex correlates with the cognitive and motivational impairments observed in schizophrenia. The prefrontal cortex is responsible for higher-order functions, including planning, problem-solving, and emotional regulation.
Neuroimaging Evidence: Patients with negative symptoms exhibit significantly lower activation in the prefrontal cortex during tasks that require executive function, further supporting the role of dopamine hypoactivity in these symptoms.
These findings underscore the significance of dopamine hypoactivity in cortical areas as a neural correlate of negative symptoms.
RESEARCH STUDIES
Davis et al. (1991) proposed the reformulated dopamine hypothesis, arguing that while positive symptoms are linked to excess dopamine in the striatum, negative symptoms are caused by dopamine underactivity in the prefrontal cortex.
Juckel et al. (2006) found that individuals with schizophrenia showed reduced dopamine activity in the prefrontal cortex, which was correlated with negative symptoms such as avolition and emotional flattening.
Weinberger et al. (1986) found that patients with prefrontal cortex damage displayed negative cognitive symptoms similar to those seen in schizophrenia. This suggests that dopamine underactivity in the prefrontal cortex plays a key role in these symptom clusters.
Goldman-Rakic et al. (2004) found that lower D1 receptor density in the prefrontal cortex was associated with cognitive deficits in schizophrenia, supporting the idea that hypodopaminergic in the prefrontal cortex contributes to working memory and decision-making impairments.
Patel et al. (2010) found that dopamine dysfunction in both the striatum and prefrontal cortex was present in patients with schizophrenia, confirming the dual role of hyperdopaminergic and hypodopaminergic activity in different pathways.
Takahashi et al. (2006) used MRI and PET scans to show that prefrontal cortex dysfunction was linked to reduced dopamine receptor availability and impaired cognitive function, further supporting the role of hypodopaminergic activity in negative symptoms.
OTHER EVIDENCE:
The new class of antipsychotics known as Atypicals, which were developed in the 1990s, support the reformulated dopamine hypothesis as they show that blocking serotonin in addition to dopamine does alleviate negative symptoms.
Atypical antipsychotics receptors, specifically block:
Dopamine D2 receptors
Serotonin 5-HT₂A receptors
This dual action is a key feature of atypical (second-generation) antipsychotics, which distinguish themselves from typical antipsychotics by:
Reducing positive symptoms (via D2 antagonism in the mesolimbic pathway)
Improving negative/cognitive symptoms and reducing side effects (via 5-HT₂A antagonism, especially in the prefrontal cortex)
At first glance, it seems illogical that blocking serotonin receptors could help with symptoms of schizophrenia that are thought to be caused by too little dopamine. If negative symptoms are linked to hyperdopaminergic in the mesocortical pathway, and drugs like risperidone and clozapine are dopamine antagonists, how do they avoid worsening this deficiency?
The answer lies in the dual mechanism of certain atypical antipsychotics. These drugs block D2 dopamine receptors — particularly in the mesolimbic pathway — to reduce positive symptoms such as hallucinations and delusions. However, they also block 5-HT₂A serotonin receptors, especially in the prefrontal cortex. This is not because serotonin itself causes schizophrenia symptoms but because 5-HT₂A activation inhibits the typical dopamine release. By blocking serotonin receptors, atypical antipsychotics remove that inhibition, enhancing dopamine release in mesocortical areas where it is abnormally low.
So it's not that serotonin is causing negative symptoms — it's that blocking serotonin allows dopamine to be restored in underactive regions. This selective enhancement is the reason second-generation antipsychotics are often better at treating negative and cognitive symptoms compared to earlier drugs, which suppress dopamine across the board.
THE REFORMULATED DOPAMINE HYPOTHESIS: WHAT IT DID NOT ADDRESS
The reformulated dopamine hypothesis was a significant improvement over the original theory. However, despite its advancements, this hypothesis still left key questions unanswered.
OTHER NEURAL CORRELATES
The dopamine hypothesis explains many symptoms of schizophrenia, but it is not the only neural correlate. Brain imaging studies have found other biological differences in people with schizophrenia, such as:
Beyond dopamine, other neural correlates have been identified in schizophrenia, including structural abnormalities. One key finding is that individuals with schizophrenia often have abnormally large ventricles, which are fluid-filled cavities in the brain. In a study, researchers compared 16 patients with "large" ventricles (more than one standard deviation above the control mean) to 16 patients with the smallest ventricles from a sample of 52 individuals with schizophrenia. They found that patients with enlarged ventricles were likelier to exhibit negative symptoms, such as alogia, affective flattening, avolition, and anhedonia. By contrast, patients with smaller ventricles tended to display positive symptoms, such as delusions, hallucinations, and bizarre behaviour. These findings suggest that combining structural imaging with symptom profiles could provide a more nuanced approach to classifying schizophrenia. However, it has been proposed that enlarged ventricles may not be a cause of the disorder but rather a result of prolonged use of antipsychotic medication, adding a layer of complexity to the interpretation of these findings.
Prefrontal cortex abnormalities – This brain area is involved in decision-making and planning. In schizophrenia, it often shows reduced activity, which may explain cognitive impairments such as poor memory and attention.
NEGATIVE SYMPTOMS ARE STILL NOT FULLY ADDRESSED
NEGATIVE SYMPTOMS AND COGNITIVE DEFICITS
The reformulated hypothesis struggled to explain negative symptoms and cognitive deficits fully. While reduced dopamine activity in the prefrontal cortex provides some answers, it is clear that dopamine alone does not account for the complexity of these symptoms. For example:
Why do negative symptoms vary so widely across patients?
Why do cognitive deficits, such as poor working memory and decision-making, persist even when dopamine levels normalise?
DELAYED THERAPEUTIC RESPONSE
Furthermore, the therapeutic delay of antipsychotics—despite their immediate blocking of dopamine receptors—indicates that dopamine likely interacts with other systems, complicating a simple cause-and-effect explanation. This suggests that dopamine imbalances interact with different neurotransmitter systems over time and that schizophrenia cannot be fully explained by dopamine alone.
MULTIPLE NEUROTRANSMITTERS
Researchers like Carlsson proposed that dopamine is just one piece of a larger neurochemical puzzle. Schizophrenia likely involves disruptions in other neurotransmitter systems, such as serotonin and glutamate. This idea is supported by the efficacy of atypical antipsychotics, such as clozapine, which targets serotonin and glutamate receptors in addition to dopamine receptors.
Carlsson did not reject the dopamine hypothesis, but he argued that it did not fully explain the symptoms of schizophrenia. His main question was: What is causing dopamine imbalances in the first place? He suggested that glutamate, another neurotransmitter, might be the key to understanding schizophrenia.
DOPAMINE IMBALANCES MAY BE A SECONDARY EFFECT
The dopamine hypothesis explains which brain areas are affected but not why dopamine is abnormal in schizophrenia.
Carlsson suggested that glutamate regulates dopamine, meaning that problems with glutamate function could be the root cause of dopamine overactivity and underactivity.
GLUTAMATE CONTROLS DOPAMINE LEVELS
Glutamate helps keep dopamine balanced in the brain.
If glutamate levels are too low, dopamine becomes unstable, leading to too much dopamine in some areas (causing psychosis) and too little in others (causing negative symptoms).
WHY DOES CLOZAPINE WORK WHEN OTHER DRUGS DON’T?
Traditional antipsychotics block D2 receptors, which help with positive symptoms but do not treat negative symptoms. Clozapine, a drug that works on both dopamine and glutamate, is effective in people who don’t respond to D2 blockers. This suggests that glutamate, not just dopamine, plays a role in schizophrenia.
EVALUATION OF ALL DOPAMINE HYPOTHESES
Overall, both the original and reformulated dopamine hypotheses shed light on the aetiology of schizophrenia, with various research methods demonstrating the link between neurotransmitter imbalances and the disorder. However, a significant question remains unresolved: the cause-and-effect dilemma. To put it simply, which came first, schizophrenia or the neurotransmitter imbalance? This "chicken-and-egg" problem raises the possibility that schizophrenia might disrupt or alter brain chemistry rather than being caused by it. Conversely, it is equally plausible that imbalances in neurotransmitter receptors, such as dopamine or glutamate, could trigger schizophrenia. Both propositions hold validity. Behaviour can also influence neurochemical changes, adding further complexity; for example, something as simple as smiling has been shown to affect serotonin production.
ALTERNATIVE BIOLOGICAL EXPLANATIONS
While the dopamine hypothesis remains influential, more recent research suggests it may be overly reductionist. Schizophrenia is increasingly viewed as a disorder involving wider neurobiological dysfunction. For example, studies have implicated glial cells, which support neuronal health and regulate inflammation in the brain. Abnormal glial function may contribute to immune dysregulation or neuroinflammatory responses, both of which are associated with schizophrenia symptomatology. This adds weight to theories that the disorder is not purely dopaminergic but may involve broader cellular and molecular disturbances.
Additionally, neurodevelopmental models propose that schizophrenia arises from early disruptions in brain development — particularly during prenatal or adolescent stages. Such disruptions may impair synaptic pruning or neural connectivity, leading to dysfunctional brain circuits long before symptoms appear. These perspectives complement dopamine-based explanations rather than replace them, highlighting the complexity of schizophrenia as a multi-factorial disorder with interacting genetic, neurochemical, and developmental components.
ISSUES AND DEBATES FOR BIOLOGICAL THEORIES - GENETIC AND NEURAL CORRELATES
DETERMINISM
All biological theories of schizophrenia are deterministic and suggest that you have no free will against developing or personally overcoming Schizophrenia. There are negative and positive aspects to this. On the plus side, parents will not be blamed for causing schizophrenia in their offspring, and individuals will not be perceived to be at fault either, as their illness is a result of their genes and/or neurotransmitters. There will, therefore, be less social stigma about being schizophrenic. However, other people may not want to procreate with schizophrenics because subsequent kids might inherit the gene.
On the negative side, excuses, excuses, excuses! Individuals and families may see it pointless to try to change their behaviour and rely on drugs to alleviate symptoms. Individuals may believe they are predestined to have Schizophrenia, which is very depressing.
PHYSIOLOGICAL REDUCTIONISM & NATURE V NURTURE
Biological explanations of schizophrenia are reductionist as they attempt to explain a complex, multi-faceted disorder at the level of genes and dopamine. Their rationale is that humans are biological organisms, and reducing even complex behaviours to neurophysiological components should be possible. As a result, biological theories overlook the importance of examining a person holistically, for example, how biology, parenting, and stress might interact as risk factors in developing the disorder.
It is now known that biology is not the only case, as only 48% of MZ twins are concurrent for schizophrenia, so psychological processes must also contribute. For example, highly expressed emotion in families has been shown to cause relapse. This demonstrates that complex phenomena cannot be easily explained by simply referring to physiological imbalance. The influence of these brain chemicals is indisputable, but to argue that they only cause schizophrenia is to neglect all other potential influences during this disorder. It may well be that, for example, stress is the ultimate cause of the disorder, creating physiological imbalances – the proximate cause.
Indeed, DSM V now believes that Schizophrenia is an aetiologically heterogeneous disorder and has thus renamed it a “spectrum” disorder. In other words, schizophrenia is a disorder that has not only a multitude of different things that can cause it, but it is also a disorder with no defining features. The addition of the term “spectrum” and the less stringent guidelines show that the DSM 5 acknowledges that it sees schizophrenia as an umbrella term and recognises that any risk factor for developing Schizophrenia will combine biology and the environment. Therefore, its cause is no longer seen as a fight between nature and nurture.
The Diathesis-Stress Model (DSM) interprets schizophrenia as a result of brain impairment in areas responsible for language and cognition. It suggests that specific brain regions, particularly those with dopamine D2 receptors, such as those in Broca's area, may be underactive or overactive. This could explain the linguistic differences in patients exhibiting positive versus negative symptoms. According to the DSM, the origins of such brain impairments in schizophrenia are multifaceted, involving a blend of genetic factors, exposure to pathogens or viruses, complications during birth, etc., all of which may interact with external stressors like abuse, bullying, or family discord.
This comprehensive perspective defines the diathesis-stress model (DS), offering a nuanced view that combines biological predispositions with environmental pressures.
SUMMARY OF THE DOPAMINE HYPOTHESES
LATE 19th CENTURY – SYNTHETIC DYE ORIGINS
Phenothiazine was first synthesised in the 1880s as part of research into coal-tar-based dyes. Its three-ring structure — containing nitrogen and sulfur — made it chemically stable and capable of binding to fabrics. It was used primarily to produce blue and violet dyes.
EARLY 20th CENTURY – CHEMICAL SIMILARITIES NOTICED
By the 1930s, researchers began to note structural similarities between phenothiazine and biologically active compounds, such as methylene blue (derived from dye research and used to treat malaria and psychosis). This sparked interest in phenothiazine derivatives as potential therapeutic agents.
1937 – PROPAZINE AND OTHER DERIVATIVES
Initial phenothiazine derivatives such as propazine were tested for various medical uses, including antihistamine and antiemetic properties, but none had profound psychiatric effects at this stage.
1940s – RISE OF ANTIHISTAMINES
As the role of histamine in allergic responses became better understood, pharmaceutical companies began exploring phenothiazine derivatives for their antihistaminic properties. This led to the synthesis of promethazine in the mid-1940s by chemists at Rhône-Poulenc.
PROMETHAZINE (1947):
Promethazine was found to have potent sedative and anti-nausea effects in addition to its antihistamine function. It was used for surgical shock, motion sickness, and pre-operative sedation. Its ability to induce calmness and indifference, even in patients who were not anxious, caught the attention of Henri Laborit, a French military surgeon.
1950 – DEVELOPMENT OF CHLORPROMAZINE
Working with Rhône-Poulenc, Laborit tested various modifications of promethazine. One derivative — chlorpromazine(RP4560) — had notably stronger sedative and emotional blunting effects. It was initially marketed as a surgical anaesthetic enhancer, but psychiatrists quickly noted its striking impact on psychotic symptoms. It reduced delusions, hallucinations, and agitation, but its mechanism of action was unknown at the time.
PSYCHIATRIC USE AND RETHINKING THE BRAIN:
By 1952, chlorpromazine was being used as the first true antipsychotic. Its effects revolutionised psychiatry and led to the dopamine hypothesis, as researchers discovered it worked by blocking dopamine receptors — even though it was derived from a dye-based antihistamine
1958–1966: EARLY RESEARCH INTO DOPAMINE AND ANTIPSYCHOTICS
Haloperidol, developed in 1958, provided further evidence that neuroleptics worked by blocking dopamine. Researchers found that both Chlorpromazine and Haloperidol were dopamine antagonists, suggesting that dopamine overactivity might play a role in psychosis — particularly positive symptoms such as hallucinations and delusions.
1971–1975: DISCOVERY OF DOPAMINE RECEPTOR SUBTYPES
Scientists identified that dopamine receptors came in different types, including D1 and D2. D2 receptors were found to be particularly important in psychosis. This led to the idea that blocking D2 receptors could reduce positive symptoms of schizophrenia.
1979–1988: THE DOPAMINE HYPOTHESIS REFINED
New research showed that not all patients with schizophrenia had elevated dopamine activity, and not all responded to dopamine-blocking drugs. This led to refinements in the dopamine hypothesis, including the idea that different brain regions may be affected differently.
1990s: A SHIFT TO REGIONAL DOPAMINE IMBALANCES
Studies found that schizophrenia might involve both hyperdopaminergic (too much dopamine) in subcortical areas like the striatum — linked to positive symptoms — and hyperdopaminergic (too little dopamine) in the prefrontal cortex — linked to negative symptoms such as emotional flattening, lack of motivation, and cognitive deficits.
1999–2010: A MORE COMPLEX VIEW OF SCHIZOPHRENIA EMERGES
Researchers began to focus on how dopamine interacts with other neurotransmitters, including glutamate and serotonin. The glutamate hypothesis gained attention after it was found that NMDA antagonists like PCP and ketamine could induce both positive and negative symptoms.
2017–2021: UNDERSTANDING SCHIZOPHRENIA AT THE MOLECULAR LEVEL
Advanced brain imaging revealed the structure and function of dopamine receptors in unprecedented detail. Researchers discovered that receptor sensitivity and distribution varied among patients, explaining differential responses to medication. Theories increasingly emphasised neurodevelopment, circuit dysfunction, and molecular heterogeneity over one-size-fits-all models
ASSESSMENT

Jay has schizophrenia. His speech is rapid and confusing, constantly changing from one idea to something completely different. Jay’s father was treated for mental health problems when he was younger. Jay’s mother worries excessively about Jay. She often criticises his behaviour and tells him what to do. Jay’s doctor prescribes medication, which seems to reduce his symptoms.
WRITING ESSAYS
Discuss one or more explanations for schizophrenia. Refer to Jay in your answer. (16 marks: AO1 – 6, A02 - 4, AO3 – 6)
WORD COUNT GUIDELINES FOR AQA A-LEVEL PSYCHOLOGY RESPONSES
6-mark A01 response: ~225 words approx.
3-mark A01 response: ~112 words approx.
SIX-MARK A01 RESPONSE (APPROX. 225 WORDS)
The dopamine hypothesis is a neural correlate of schizophrenia, as it identifies dopamine imbalances in specific brain regions as a cause of symptoms. The original dopamine hypothesis suggested that positive symptoms, such as hallucinations and delusions, result from excess dopamine (hyperdopaminergic activity) in the mesolimbic pathway. This pathway connects the ventral tegmental area (VTA) to the nucleus accumbens, a structure involved in reward and motivation. Overactivation of D2 receptors in this system disrupts standard information processing, leading to distorted perception and thought.
However, this model did not explain negative symptoms, such as apathy and cognitive dysfunction, leading to the reformulated dopamine hypothesis. This version proposes that negative symptoms arise due to too little dopamine (hypodopaminergic activity) in the mesocortical pathway. This connects the VTA to the prefrontal cortex, a region responsible for higher-order thinking, planning, and motivation. D1 receptors in the prefrontal cortex are excitatory, meaning they enhance brain activity, but dopamine underactivity in this pathway reduces prefrontal function, leading to persistent negative symptoms.
Since distinct patterns of dopamine dysfunction correspond to different symptom types, the dopamine hypothesis provides a biological explanation for schizophrenia. It is considered a neural correlate because it links specific brain abnormalities to the development of symptoms, helping to explain why schizophrenia presents with both excessive and diminished mental activity.
THREE-MARK A01 RESPONSE (APPROX. 112 WORDS)
The dopamine hypothesis is a neural correlate of schizophrenia, as it links dopamine dysfunction in specific brain pathways to distinct symptoms. The original dopamine hypothesis suggested that positive symptoms, such as hallucinations and delusions, result from too much dopamine (hyperdopaminergic activity) in the mesolimbic pathway. This system connects the ventral tegmental area (VTA) to the nucleus accumbens, which processes reward and emotion. Overactivation of D2 receptors in this system disrupts thought processing, leading to perceptual distortions.
The reformulated dopamine hypothesis explains negative symptoms, such as apathy, by suggesting that they result from too little dopamine (hypodopaminergic activity) in the mesocortical pathway. This pathway links the VTA to the prefrontal cortex, which controls motivation and cognitive function. D1 receptors in this region require sufficient dopamine, and when levels are too low, negative symptoms such as reduced motivation and cognitive impairment emerge.
WRITING A03
RESEARCH ANALYSIS: LEGAL DOPAMINE ANTAGONISTS (ANTIPSYCHOTIC MEDICATIONS)
Strengths:
The most substantial empirical support for the dopamine hypothesis comes from the success of typical antipsychotic drugs (e.g., chlorpromazine) in reducing positive symptoms of schizophrenia. These drugs block dopamine D2 receptors, reducing dopamine transmission in the mesolimbic pathway.
Atypical antipsychotics (e.g., clozapine, risperidone) are also effective, and while they target dopamine, they additionally affect serotonin (5-HT2A receptors), which helps alleviate negative symptoms such as avolition and affective flattening.
The effectiveness of these medications directly supports the idea that excess dopamine contributes to psychotic symptoms, as reducing dopamine levels alleviates hallucinations and delusions.
Weaknesses:
While typical antipsychotics support the dopamine hypothesis by reducing positive symptoms, they fail to address negative symptoms like social withdrawal and cognitive dysfunction. This suggests dopamine alone cannot fully explain schizophrenia.
Some patients with schizophrenia do not respond to dopamine antagonists, implying other neurotransmitter systems (e.g., glutamate, serotonin) contribute to the disorder.
Dopamine dysfunction does not account for all neural correlates of schizophrenia. Brain imaging studies suggest structural abnormalities (e.g., enlarged ventricles, prefrontal cortex dysfunction) are also involved, indicating that schizophrenia is not just a chemical imbalance but a disorder with broader neurobiological underpinnings.
Conclusion:
The efficacy of dopamine-blocking drugs in treating positive symptoms supports the dopamine hypothesis. Still, their limited effect on negative symptoms and individual differences in response suggest additional factors contribute to schizophrenia.
The fact that atypical antipsychotics also target serotonin and show greater effectiveness in treating negative symptoms highlights the need for a broader neurotransmitter model beyond dopamine alone.
RESEARCH ANALYSIS: ANIMAL STUDIES (RATS)
Strengths: Rats share a similar mesolimbic dopamine system with humans, making them valuable for studying the effects of dopamine agonists and antagonists.
Weaknesses: Rats lack human-specific cognitive functions, particularly language, central to schizophrenia. The inability to directly assess hallucinations or delusions in rats raises validity concerns. Furthermore, schizophrenia may be uniquely human, limiting generalisability.
Conclusion: While rat studies provide insight into dopamine pathways, their inability to replicate the full human experience of schizophrenia weakens their applicability.
RESEARCH ANALYSIS: ANTIPSYCHOTICS & PARKINSON'S DISEASE
Strengths: The link between Parkinson’s disease and schizophrenia provides compelling evidence for dopamine’s role. L-Dopa (a dopamine agonist) can induce psychotic symptoms in Parkinson’s patients, mirroring schizophrenia. Conversely, dopamine antagonists used in schizophrenia treatment can cause Parkinsonian symptoms.
Weaknesses: The exact mechanisms of schizophrenia differ from drug-induced psychosis. Parkinson’s patients do not develop all schizophrenia symptoms, suggesting additional neurochemical or structural abnormalities.
Conclusion: The Parkinson’s-schizophrenia link supports the dopamine hypothesis but does not account for all symptoms or underlying causes.
RESEARCH ANALYSIS: ILLEGAL DRUGS
Strengths: Drug-induced psychosis mimics schizophrenia symptoms, supporting the role of dopamine excess in positive symptoms. Studies from the 1970s induced amphetamine psychosis to examine schizophrenia-like behaviours, reinforcing the dopamine hypothesis.
Weaknesses: Drug-induced psychosis is now considered qualitatively different from schizophrenia, with additional symptoms like euphoria and hyperactivity resembling mania rather than schizophrenia.
Conclusion: While illicit drug studies reinforce dopamine’s role in psychosis, they cannot fully explain schizophrenia’s complexity.
POST-MORTEM STUDIES
Strengths: Early post-mortem studies found increased dopamine receptor density, suggesting a biological basis for schizophrenia.
Weaknesses: Post-mortem brains often belonged to medicated individuals, meaning receptor density changes could result from long-term drug use rather than schizophrenia itself. These studies also lack real-time dopamine activity data.
Conclusion: While post-mortem studies provided early evidence for the dopamine hypothesis, their methodological limitations make modern imaging techniques more reliable.
BRAIN IMAGING: POSITIVE SYMPTOMS
Strengths: PET and fMRI studies have confirmed hyperdopaminergic activity in schizophrenia. Increased dopamine receptor density in the striatum correlates with psychotic symptoms.
Key Studies:
Lindström et al. (1999): Increased dopamine synthesis in the striatum of schizophrenic patients.
Howes et al. (2012): Elevated dopamine synthesis in individuals at high risk of schizophrenia, supporting dopamine's causal role.
Laruelle et al. (1996): Amphetamine challenge studies show greater dopamine release in schizophrenics than controls.
Weaknesses: Correlational nature—dopamine imbalances may be a consequence, not a cause, of schizophrenia.
Conclusion: Strong evidence links dopamine excess in the mesolimbic pathway to positive symptoms, but causation remains uncertain.
BRAIN IMAGING: NEGATIVE SYMPTOMS
Strengths: Negative symptoms correlate with dopamine hypoactivity in cortical areas, particularly:
Ventral striatum: Reduced activation linked to avolition.
Prefrontal cortex: Lower dopamine D1 receptor function associated with cognitive deficits.
Weaknesses: Imaging studies cannot establish causation—dopamine dysfunction could be a result rather than a cause.
Conclusion: Dopamine hypoactivity in cortical areas likely underlies negative symptoms, supporting a more nuanced dopamine hypothesis.
BEYOND DOPAMINE: GLUTAMATE & OTHER NEUROTRANSMITTERS
Glutamate's Role: Carlsson suggested dopamine imbalances may result from glutamate dysfunction. Glutamate regulates dopamine, and its disruption can cause both hyperdopaminergic (positive symptoms) and hypodopaminergic (negative symptoms) states.
Supporting Evidence: Clozapine, an effective antipsychotic for treatment-resistant schizophrenia, affects both dopamine and glutamate, suggesting multiple neurotransmitter involvement.
Conclusion: The dopamine hypothesis alone is insufficient—glutamate and serotonin likely play crucial roles in schizophrenia.
DOPAMINE AND OTHER PSYCHIATRIC DISORDERS
Dopamine imbalances are not unique to schizophrenia and are implicated in other psychiatric conditions, reinforcing the idea that dopamine dysfunction alone is insufficient to explain schizophrenia.
Bipolar Disorder (Mania): Elevated dopamine activity is associated with manic episodes, supporting the idea that dopamine dysregulation can lead to psychotic symptoms beyond schizophrenia.
Acute and Transient Psychotic Disorder: This disorder presents with brief episodes of psychosis linked to dopamine fluctuations, but unlike schizophrenia, it lacks persistent cognitive and negative symptoms.
Schizoaffective Disorder: This disorder has both schizophrenic and mood disorder symptoms, indicating dopamine dysfunction interacts with serotonin and glutamate imbalances.
Parkinson’s Disease and Dopamine Agonists: Parkinson’s is caused by dopamine deficiency in the substantia nigra. When treated with dopamine agonists (e.g., L-Dopa), some patients develop hallucinations and delusions, supporting the link between dopamine excess and psychotic symptoms.
CONCLUSION:
Dopamine imbalances contribute to multiple psychiatric conditions, demonstrating that dopamine dysfunction is not specific to schizophrenia.
The fact that schizophrenia shares dopamine abnormalities with other disorders suggests additional neural mechanisms must be involved, such as glutamate dysfunction, structural abnormalities, and prefrontal cortex deficits.
The dopamine hypothesis remains central to understanding schizophrenia but must be considered alongside other neural correlates and neurotransmitter theories.
ISSUES & DEBATES:
DETERMINISM
Positive: Removes blame from individuals and families.
Negative: This could lead to fatalism, discouraging behavioural interventions.
Conclusion: While a deterministic view has benefits, it risks reinforcing helplessness.
REDUCTIONISM
Positive: Identifying biological mechanisms allows for targeted treatments.
Negative: Oversimplifies schizophrenia by ignoring environmental and psychological factors (e.g., stress, trauma).
Conclusion: Integrating biological and environmental factors, a diathesis-stress model offers a more comprehensive explanation.
FINAL EVALUATION
Strengths of the Dopamine Hypothesis:
Supported by drug studies, imaging, and antipsychotic efficacy.
Explains positive and negative symptoms through differential dopamine activity.
Weaknesses:
Causality issue—dopamine imbalances may be a consequence rather than a cause.
It cannot fully explain schizophrenia’s complexity—other neurotransmitters are involved.
Structural abnormalities (e.g., enlarged ventricles) suggest a broader neuropathological basis.
Conclusion: The dopamine hypothesis remains a cornerstone of schizophrenia research but must be integrated with newer models considering glutamate, serotonin, and structural abnormalities.